
Doença renal policística autossômica dominante - Autosomal dominant polycystic kidney disease
| Doença renal policística autossômica dominante | |
|---|---|
| Outros nomes | PKD autossômica dominante, PKD de início adulto |
 | |
| Rins policísticos | |
| Especialidade |
Genética Médica |
A doença renal policística autossômica dominante ( ADPKD ) é a doença monogênica humana mais prevalente e potencialmente letal . Está associada a grande variabilidade interfamiliar e intrafamiliar, que pode ser explicada em grande parte por sua heterogeneidade genética e genes modificadores . É também a mais comum das doenças renais císticas hereditárias - um grupo de doenças com patogênese relacionada, mas distinta, caracterizada pelo desenvolvimento de cistos renais e várias manifestações extra-renais, que no caso de ADPKD incluem cistos em outros órgãos, como o fígado , vesículas seminais , pâncreas e membrana aracnóide , bem como outras anormalidades, como aneurismas intracranianos e dolicoectasias , dilatação da raiz aórtica e aneurismas, prolapso da válvula mitral e hérnias da parede abdominal . Mais de 50% dos pacientes com ADPKD eventualmente desenvolvem doença renal em estágio terminal e requerem diálise ou transplante renal . Estima-se que a ADPKD afete pelo menos um em cada 1000 indivíduos em todo o mundo, tornando esta doença a doença renal hereditária mais comum, com uma prevalência diagnosticada de 1: 2000 e incidência de 1: 3000-1: 8000 em escala global.
sinais e sintomas
Entre as apresentações clínicas estão:
- Dor aguda no lombo
- Sangue na urina
- Rins votáveis
- Hemorragia subaracnóide (aneurisma da baga)
- Hipertensão
- Cistos hepáticos associados
- Uremia devido a insuficiência renal
- Anemia devido a doença renal crônica
- Aumentar a secreção de hemácias ou eritropoietina
Genética
ADPKD é geneticamente heterogêneo com dois genes identificados: PKD1 (região cromossômica 16p13.3; cerca de 85% dos casos) e PKD2 (4q21; cerca de 15% dos casos). Vários mecanismos genéticos provavelmente contribuem para a expressão fenotípica da doença. Embora existam evidências de um mecanismo de dois acertos (linha germinativa e inativação somática de dois alelos PKD) explicando o desenvolvimento focal de cistos renais e hepáticos, é mais provável que a haploinsuficiência seja responsável pelas manifestações vasculares da doença. Além disso, os novos modelos de ratinhos homozigóticos para PKD1 alelos hipomórfico 22 e 23 e a demonstração de aumento da proliferação celular epitelial renal em PKD2 +/- ratinhos sugerem que os outros do que a hipótese de dois mecanismos de acerto também contribuir para o fenótipo cística.
Grande variabilidade interfamiliar e intrafamiliar ocorre na ADPKD. A maioria dos indivíduos com mutações PKD1 tem insuficiência renal aos 70 anos, enquanto mais de 50% dos indivíduos com mutações PKD2 têm função renal adequada nessa idade (idade média de início da doença renal em estágio terminal: 54,3 anos com PKD1 ; 74 · 0 anos com PKD2 ).
A variabilidade intrafamiliar significativa observada na gravidade das manifestações renais e extra-renais aponta para fatores modificadores genéticos e ambientais que podem influenciar o resultado de ADPKD, e os resultados de uma análise da variabilidade na função renal entre gêmeos monozigóticos e irmãos apoiam o papel dos modificadores genéticos nesta doença. Estima-se que 43-78% da variação na idade para ESRD pode ser devido a fatores modificadores hereditários, com os pais tão propensos quanto as crianças a apresentarem doença mais grave em estudos de pares de pais e filhos.
Fisiopatologia
Em muitos pacientes com ADPKD, a disfunção renal não é clinicamente aparente até os 40 ou 50 anos de vida. No entanto, um crescente corpo de evidências sugere que a formação de cistos renais começa no útero . Os cistos inicialmente se formam como pequenas dilatações nos túbulos renais, que então se expandem para formar cavidades cheias de líquido de diferentes tamanhos. Os fatores sugeridos para levar à cistogênese incluem uma mutação da linha germinativa em um dos alelos do gene da policistina, um segundo acerto somático que leva à perda do alelo normal e um terceiro acerto, que pode ser um insulto renal que desencadeia a proliferação celular e um resposta a lesões. . Devido a inúmeras semelhanças entre a fisiopatologia da ADPKD e a fisiopatologia da resposta renal à lesão, a ADPKD foi descrita como um estado de ativação aberrante e persistente das vias de resposta à lesão renal. Na progressão da doença, a dilatação contínua dos túbulos por meio do aumento da proliferação celular, secreção de fluido e separação do túbulo parental leva à formação de cistos.
A ADPKD, junto com muitas outras doenças que se apresentam com cistos renais, pode ser classificada em uma família de doenças conhecidas como ciliopatias . As células epiteliais dos túbulos renais, incluindo todos os segmentos do néfron e os ductos coletores (com exceção das células intercaladas), mostram a presença de um único cílio apical primário. A policistina -1 , a proteína codificada pelo gene PKD1 , está presente nesses cílios e acredita-se que detecte o fluxo com seus grandes domínios extracelulares, ativando os canais de cálcio associados à policistina-2 , produto do gene PKD2 , como resultado de a configuração genética de ADPKD conforme explicado na subseção genética acima.
A proliferação de células epiteliais e a secreção de fluido que levam à cistogênese são duas características marcantes na ADPKD. Durante os estágios iniciais da cistogênese, os cistos são fixados em seus túbulos renais parentais e um derivado do filtrado glomerular entra nos cistos. Uma vez que esses cistos se expandem para aproximadamente 2 mm de diâmetro, o cisto se fecha de seu túbulo parental e depois que o líquido só pode entrar nos cistos por meio da secreção transepitelial, que por sua vez é sugerida a aumentar devido aos efeitos secundários de um aumento da concentração intracelular de cíclico AMP (cAMP).
Clinicamente, o aumento insidioso do número e do tamanho dos cistos renais se traduz em aumento progressivo do volume renal. Estudos conduzidos por profissionais da Mayo Clinic estabeleceram que o volume renal total (TKV) em uma grande coorte de pacientes com ADPKD foi de 1.060 ± 642ml com um aumento médio de 204ml em três anos, ou 5,27% ao ano no curso natural da doença, entre outras descobertas novas e importantes que foram amplamente estudadas pela primeira vez.

Diagnóstico
Normalmente, o diagnóstico de ADPKD é realizado inicialmente por imagens renais usando ultrassonografia , tomografia computadorizada ou ressonância magnética . No entanto, o diagnóstico molecular pode ser necessário nas seguintes situações: 1- quando um diagnóstico definitivo é necessário em indivíduos jovens, como um potencial doador vivo parente em uma família afetada com dados de imagem duvidosos; 2- em pacientes com história familiar negativa de ADPKD, devido à potencial sobreposição fenotípica com várias outras doenças císticas renais; 3- em famílias com doença renal policística de início precoce, pois nesses casos alelos hipomórficos e / ou herança oligogênica podem estar envolvidos; e 4- em pacientes que solicitam aconselhamento genético , especialmente em casais que desejam um diagnóstico genético pré-implantação .
Os achados de grandes rins ecogênicos sem cistos macroscópicos distintos em um lactente / criança com risco de 50% de ADPKD são diagnósticos. Na ausência de história familiar de ADPKD, a presença de aumento renal bilateral e cistos, com ou sem a presença de cistos hepáticos , e a ausência de outras manifestações sugestivas de uma doença cística renal diferente fornecem evidências presumivelmente, mas não definitivas, de o diagnóstico. Em alguns casos, aneurismas intracranianos podem ser um sinal associado de ADPKD, e o rastreamento pode ser recomendado para pacientes com história familiar de aneurisma intracraniano.
O teste genético molecular por análise de ligação ou triagem de mutação direta está clinicamente disponível; no entanto, a heterogeneidade genética é uma complicação significativa para os testes de genética molecular . Às vezes, um número relativamente grande de membros da família afetados precisa ser testado para estabelecer qual dos dois genes possíveis é o responsável em cada família. O grande tamanho e complexidade de PKD1 e PKD2 genes , assim como marcados heterogeneidade alélica , constituem obstáculos ao teste molecular por directo análise de ADN . A sensibilidade do teste é de quase 100% para todos os pacientes com ADPKD com 30 anos ou mais e para pacientes mais jovens com mutações PKD1 ; esses critérios são apenas 67% sensíveis para pacientes com mutações PKD2 ]] que têm menos de 30 anos.

Rim policístico adulto

Diagrama de doença policística autossômica dominante com rim normal inserido para comparação
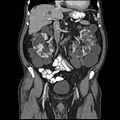
Tomografia computadorizada abdominal de um adulto com doença renal policística autossômica dominante: A formação extensa de cistos é observada em ambos os rins, com alguns cistos no fígado também. ( Plano coronal )



Tratamento
Atualmente, o único tratamento farmacológico disponível para ADPKD consiste na redução da velocidade de ganho do volume renal total (TKV) com antagonistas do receptor 2 da vasopressina (V2) (isto é, tolvaptano). O tratamento com tolvaptan não interrompe ou reverte a progressão da doença e os pacientes ainda progridem para a insuficiência renal. As modalidades de tratamento paliativo envolvem medicamentos sintomáticos (analgésicos não opioides e opioides) para dor abdominal / retroperitoneal. As opções para dor resistente a analgésicos incluem procedimentos cirúrgicos simples ou complexos (ou seja, aspiração do cisto renal, decorticação do cisto, denervação renal e nefrectomia), que podem resultar em complicações inerentes à cirurgia. Pesquisas recentes sugerem que as intervenções dietéticas cetogênicas afetam beneficamente a progressão e os sintomas em indivíduos com ADPKD. A perda leve de peso afeta favoravelmente a dor, indicando o benefício de mudanças na dieta e no estilo de vida.
Medicação aquarética
Em 2014, o Japão foi o primeiro país do mundo a aprovar um tratamento farmacológico para ADPKD, seguido pelo Canadá e Europa, que aprovaram o medicamento tolvaptano para pacientes com ADPKD no início de 2015. O FDA dos EUA aprovou o uso de tolvaptano no tratamento de ADPKD em 2018. Tolvaptan, uma droga aquarética , é um antagonista do receptor 2 da vasopressina (V2) . Estudos pré-clínicos sugeriram que a molécula cAMP poderia estar envolvida no aumento dos cistos ADPKD, e estudos em roedores confirmaram o papel da vasopressina no aumento dos níveis de cAMP no rim, o que lançou a base para a condução de estudos clínicos. Porque os dados do Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) liderado pela Mayo Clinic mostraram que o volume renal total (TKV) previu o risco de desenvolver doença renal crônica em pacientes com ADPKD, o ensaio TEMPO 3: 4, que inscreveu pacientes de 129 locais em todo o mundo de 2007 a 2009, avaliaram o TKV como um ponto final primário para testar a eficácia do tolvaptano em pacientes com ADPKD. Esse estudo mostrou uma diminuição significativa na proporção de aumento de TKV e dissuasão do declínio da função renal em pacientes com ADPKD após o tratamento com tolvaptan; entretanto, como os resultados dos exames laboratoriais relativos à função hepática pareceram elevados em uma porcentagem de pacientes inscritos naquele estudo, a aprovação do medicamento foi adiada pelas agências regulatórias ou, como no caso dos Estados Unidos, totalmente negada.
Intervenções dietéticas e de estilo de vida
Pesquisas usando modelos de camundongos ADPKD mostraram que a restrição alimentar moderada melhorou fortemente a progressão da doença. Foi demonstrado que o mecanismo envolve o estado metabólico de cetose, e os efeitos benéficos podem ser produzidos por alimentação restrita por tempo, jejum agudo, dieta cetogênica ou por suplementação com beta-hidroxibutirato de cetona em modelos de camundongo, rato e gato de ADPKD. Um regime de dieta cetogênica não apenas interrompeu a progressão da doença, mas levou à reversão parcial da doença cística renal em um modelo de rato. O estado metabólico da cetose pode ser benéfico no ADPKD porque as células do cisto renal no ADPKD têm um defeito metabólico semelhante ao efeito Warburg no câncer que as torna altamente dependentes da glicose e incapazes de metabolizar ácidos graxos e cetonas. Consistente com isso, os níveis de glicose sérica se correlacionam positivamente com a progressão mais rápida da doença em pacientes com ADPKD. Além disso, os indivíduos com ADPKD e diabetes tipo 2 têm volume renal total (TKV) significativamente maior do que aqueles com ADPKD sozinho, e o sobrepeso ou a obesidade estão associados a uma progressão mais rápida no estágio inicial de ADPKD. Um estudo retrospectivo de série de casos mostrou que os sintomas da doença ADPKD - incluindo dor, hipertensão e função renal - melhoraram entre 131 pacientes que implementaram dietas cetogênicas por uma duração média de 6 meses.
A ingestão dietética de sódio está associada a pior declínio da função renal na ADPKD, e limitar a ingestão de sódio é geralmente recomendado aos pacientes. A ingestão de proteína dietética não se correlacionou com a progressão da ADPKD.
Acredita-se que o aumento da ingestão de água seja benéfico no ADPKD e geralmente é recomendado. O mecanismo benéfico subjacente do aumento da ingestão de água pode estar relacionado aos efeitos sobre o receptor V2 da vasopressina ou pode ser devido à supressão da formação de microcristais prejudiciais nos túbulos renais por diluição de solutos como oxalato de cálcio, fosfato de cálcio e ácido úrico.
Medicação analgésica
A dor crônica em pacientes com DRPAD geralmente é refratária a tratamentos conservadores e não invasivos, mas analgésicos não opioides e intervenções conservadoras podem ser usados antes que os analgésicos opioides sejam considerados; se a dor continuar, as intervenções cirúrgicas podem ter como alvo os cistos renais ou hepáticos para abordar diretamente a causa da dor, com opções cirúrgicas incluindo decorticação do cisto renal , denervação renal e nefrectomia .
Aspiração de cisto renal
A aspiração com escleroterapia com etanol pode ser realizada para o tratamento de cistos renais simples sintomáticos, mas pode ser impraticável em pacientes avançados com cistos múltiplos. O procedimento em si consiste na inserção percutânea de uma agulha no cisto identificado, sob orientação de ultrassom , com posterior drenagem do líquido contido; a escleroterapia é usada para evitar a reacumulação de líquido que pode ocorrer no cisto, o que pode resultar na recorrência dos sintomas.
Decorticação de cisto laparoscópico
A decorticação laparoscópica do cisto (também conhecida como marsupialização) consiste na remoção de um ou mais cistos renais por meio de cirurgia laparoscópica , durante a qual os cistos são puncionados e a parede externa dos cistos maiores é excisada com cuidado para não incisar o parênquima renal. Esse procedimento pode ser útil para o alívio da dor em pacientes com DRPAD e geralmente é indicado após a aspiração precoce do cisto confirmar que o cisto a ser decorticado é responsável pela dor. Ensaios clínicos controlados não randomizados realizados na década de 90 mostraram que pacientes com cistos renais simples sintomáticos que apresentavam recorrência dos sintomas após resposta inicial à aspiração simples podiam ser submetidos à decorticação do cisto com segurança, com vida média sem dor entre 17 e 24 meses após a cirurgia. A decorticação laparoscópica apresenta uma taxa de recorrência de 5% dos cistos renais em comparação com uma taxa de recorrência de 82% obtida com a escleroterapia.
Neurólise
Um novo tratamento especificamente para a dor crônica sofrida por muitos portadores de ADPKD é a neurólise do plexo celíaco . Isso envolve a ablação química do plexo celíaco , para causar uma degeneração temporária das fibras nervosas visadas. Quando as fibras nervosas se degeneram, isso causa uma interrupção na transmissão dos sinais nervosos. Esse tratamento, quando bem-sucedido, proporciona alívio significativo da dor por um período que varia de alguns dias a mais de um ano. O procedimento pode ser repetido quando os nervos afetados estiverem curados e a dor voltar.
Nefrectomia
Muitos pacientes com ADPKD sofrem sequelas sintomáticas em consequência da doença, como hemorragia de cisto , dor no flanco , infecções recorrentes , nefrolitíase e sintomas de efeito de massa (isto é, saciedade precoce , náuseas e vômitos e desconforto abdominal), de seus rins aumentados. Em tais casos, a nefrectomia pode ser necessária devido a sintomas intratáveis ou quando no decurso da preparação para o transplante de rim , os rins nativos colidem com a pelve verdadeira e impedem a colocação de um aloenxerto de doador . Além disso, a nefrectomia nativa pode ser realizada na presença de suspeita de malignidade, uma vez que o carcinoma de células renais (RCC) é duas a três vezes mais provável na população com DRPAD em doença renal em estágio terminal (ESKD) do que nos pacientes com DRCT sem DRPAD. Embora as indicações para nefrectomia em ADPKD possam estar relacionadas ao tamanho do rim, a decisão de prosseguir com a nefrectomia nativa é frequentemente realizada individualmente, sem referência específica às medições do tamanho do rim.
Diálise
Duas modalidades de diálise podem ser usadas no tratamento de pacientes com DRPAD: diálise peritoneal e hemodiálise . Dados epidemiológicos mostram que ADPKD afeta 5-13,4% dos pacientes em hemodiálise na Europa e nos Estados Unidos, e cerca de 3% no Japão. A diálise peritoneal geralmente tem sido contra-indicada em pacientes com DRPAD com grandes volumes renais e hepáticos, devido às dificuldades físicas esperadas no procedimento e possíveis complicações; no entanto, nenhuma diferença é observada na morbidade a longo prazo entre a hemodiálise e a diálise peritoneal em ADPKD.
Transplante de rim
O transplante renal é aceito como o tratamento preferencial para pacientes com DRPAD com ESRD. Entre os pacientes americanos na lista de espera para transplante renal (em dezembro de 2011), 7.256 (8,4%) foram listados devido à doença renal cística e dos 16.055 transplantes renais realizados em 2011, 2.057 (12,8%) foram feitos para pacientes com doença cística doença renal, com 1.189 de doadores falecidos e 868 de doadores vivos.
Prognóstico
Em pacientes com ADPKD, o desenvolvimento e a expansão graduais do cisto resultam no aumento do rim e, durante o curso da doença, a taxa de filtração glomerular permanece normal por décadas antes que a função renal comece a deteriorar-se progressivamente, dificultando a previsão precoce do resultado renal. O estudo CRISP, mencionado na seção de tratamento acima, contribuiu para construir uma base lógica sólida que apóia o valor prognóstico do volume renal total (TKV) em ADPKD; O TKV (avaliado por ressonância magnética ) aumenta de forma constante e uma maior taxa de aumento renal se correlacionou com o declínio acelerado da TFG, enquanto o TKV ajustado pela altura do paciente (HtTKV) ≥600 ml / m prediz o desenvolvimento de doença renal crônica estágio 3 em 8 anos.
Além do TKV e do HtTKV, a taxa de filtração glomerular estimada (eTFG) também foi usada provisoriamente para prever a progressão da ADPKD. Após a análise de TC ou ressonância magnética de 590 pacientes com ADPKD tratados no Mayo Translational Polycystic Kidney Disease Center , Irazabal e colegas desenvolveram um sistema de classificação baseado em imagem para prever a taxa de declínio de eGFR em pacientes com ADPKD. Neste método de prognóstico, os pacientes são divididos em cinco subclasses de taxas de crescimento renal estimadas de acordo com intervalos de HtTKV específicos para a idade (1A, <1,5%; 1B, 1,5–3,0%; 1C, 3,0–4,5%; 1D, 4,5–6,0% ; e 1E,> 6,0%) conforme delineado no estudo CRISP. O declínio na eTFG ao longo dos anos após a medição inicial de TKV é significativamente diferente entre todas as cinco subclasses de pacientes, com aqueles na subclasse 1E tendo o declínio mais rápido. Algumas das causas mais comuns de morte em pacientes com ADPKD são várias infecções (25%), um aneurisma de baga rompido (15%) ou doença cardíaca coronária / hipertensiva (40%).
Referências
links externos
- https://web.archive.org/web/20110608142128/http://kidney.niddk.nih.gov/kudiseases/pubs/polycystic/index.htm
- https://www.ncbi.nlm.nih.gov/disease/PKD.html
| Classificação | |
|---|---|
| Fontes externas |